
TALASEMIAS: LAS ANEMIAS HEREDADAS
Las talasemias son un conjunto heterogéneo de trastornos hereditarios de la sangre. Todas ellas derivan de la síntesis insuficiente o anómala de hemoglobina y cursan como procesos anémicos.
Se conocen varios tipos de talasemias y se clasifican dependiendo de las cadenas de la hemoglobina que estén afectadas, así como de la gravedad con que se desarrolla la enfermedad.
ESTRUCTURA DE LA HEMOGLOBINA Y TIPOS DE TALASEMIAS
La hemoglobina es una de las proteínas que conforman los glóbulos rojos y la responsable de que estos puedan transportar oxígeno unido a ella (oxihemoglobina) de los pulmones a los tejidos a través del torrente sanguíneo y haciendo lo propio con el dióxido de carbono (carboxihemoglobina) originado en las células, trasportándolo a los pulmones para ser expulsado.
La molécula está formada por 4 subunidades de naturaleza proteica llamadas globinas: 2 alfa (α) y 2 beta (β). Las alteraciones estructurales de la hemoglobina afectan a la eficiencia de unión y transporte de las moléculas de oxígeno.
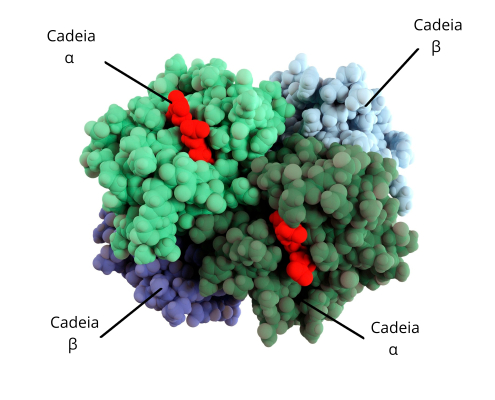
Las talasemias se deben a defectos en la síntesis de las cadenas alfa o beta de la hemoglobina como consecuencia de mutaciones en los genes responsables de su codificación. Ese defecto lleva a la acumulación de globina en el hematíe destruyéndolo y causando la anemia.
Se engloban en dos grandes grupos en función del tipo de cadena dañada:
-Las alfa-talasemias, derivadas de la alteración en los genes que codifican para la proteína alfa-globina de la hemoglobina.
-Las beta-talasemias, en las que están mutados los genes responsables de la formación de la cadena beta de la hemoglobina (proteína beta-globina).
| α-TALASEMIAS | Β-TALASEMIAS | |||||
| Causadas por deleciones o mutaciones puntuales en los genes de alfa-globina: HBA1, HBA2, ambos en el cromosoma 16
A mayor número de alelos afectados, más grave es la enfermedad
|
Causadas la deficiencia o ausencia de síntesis
de cadenas β de globina Afectado el gen HBB, en el cromosoma 11 Exceso de cadenas α de globina precipitan y dañan los hematíes provocando la anemia por hemólisis. |
|||||
| α-talasemia
(portador silente) |
α-talasemia menor
(rasgo talasémico) |
Hemoglobina H
(talasemia intermedia) |
α-talasemia mayor | δβ-talasemia homocigota
(β-talasemia intermedia) |
β-talasemia menor
(rasgo talasémico) |
β-talasemia mayor
(Anemia de Cooley) |
| Afectado un gen de la alga-globina
Suele ser asintomático |
Dos genes de la alfa-globina afectados
Anemia leve
|
Tres genes de la alfa-globina afectados
Anemia moderada Requiere tratamiento Autosómica Recesiva |
Cuatro genes de la alfa-globina afectados
Normalmente incompatible con la vida |
Afecta a dos genes de la beta-globina
Anemia moderada No suele requerir transfusiones Autosómica Dominante |
Un gen de la beta-globina afectado
Asintomática |
Afecta a dos genes de la beta-globina
Anemia severa Suele requerir transfusiones de sangre y otras intervenciones |
TALASEMIAS Y HERENCIA
Las talasemias siguen fundamentalmente un patrón de herencia autosómico recesivo (siendo algunas formas como la beta-talasemia intermedia autosómicas dominantes), es decir, los hijos de dos portadores sanos tienen una probabilidad de un 25% de manifestar la enfermedad, ya sea moderadas o severa, dependiendo del tipo de mutación que se transmita.
Ejemplo de patrón de herencia de talasemia autosómico recesivo:
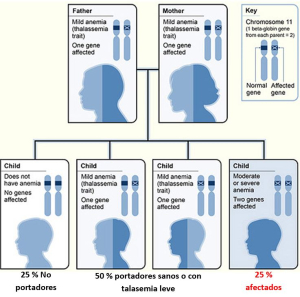
En cualquier caso, presentando una forma leve e incluso asintomática de la enfermedad, esta puede transmitirse a la descendencia. Por ello, y dado que no hay ningún modo de prevenirla, lo recomendable es que las personas afectadas recurran al consejo genético antes de tener hijos.
¿CÓMO SABER SI TENGO TALASEMIA?
El síntoma más característico de las talasemias es la anemia. Esta cursa con síntomas como el cansancio, debilidad, dificultad para respirar, piel pálida, mareo, etc.
Los casos leves no presentan síntomas, pero las formas graves o moderadas son diagnosticadas mediante analíticas de sangre en las que se incluya hemograma, proteinograma o estudio genético.
Asimismo, los casos graves pueden tener otros síntomas derivados de la degradación de los hematíes. Estos incluyen ictericia por sobrecarga del hígado, hipertrofia del bazo, hipertrofia de la médula ósea, debilidad en los huesos, etc.
La anemia sostenida incrementa la absorción de hierro como medida homeostática compensatoria. Esta, unida a las transfusiones habituales, implica un exceso de hierro que puede depositarse en el músculo cardiaco y provocar su insuficiencia.
TRATAMIENTO
Las talasemias no pueden curarse ni prevenirse al ser una enfermedad genética. Si bien, dependiendo de la gravedad de la variante presentada, es posible paliar los síntomas en mayor o menor medida.
Los casos muy graves resultan incompatibles con la vida, como en el caso de la alfa-talasemia mayor. Por su parte, los leves y moderados son tratados con diferentes abordajes enfocados a compensar la anemia derivada de la degradación de los hematíes dañados.
Estos pueden ser:
-En talasemias graves o moderadas se requieren transfusiones regulares que aumenten el hematocrito de la persona afectada, mejorando el transporte de oxígeno y aliviando lo síntomas de la anemia.
-Terapia de quelación del hierro. Como ya se ha mencionado, el aumento de absorción de hierro como consecuencia de la anemia, unido a las frecuentes transfusiones, lleva a un exceso de hierro en sangre que se acumula en órganos como corazón o el hígado. Los medicamentos quelantes del hierro se unen a él para que pueda ser eliminado.
-Suplementos nutricionales.
-La hiperplasia del bazo puede hacer necesaria su extirpación.
-En casos graves el trasplante de médula ósea es una opción curativa. Este procedimiento implica la sustitución de las células de la médula ósea que fabrican globinas “defectuosas” por células sanas de un donante compatible. No obstante, el trasplante de médula ósea tiene sus propios riesgos y no siempre es una opción adecuada.
-Terapia génica. Este tratamiento está aún en investigación y consiste en corregir los genes mutados responsables de la enfermedad.
Cada paciente aquejado de talasemia deberá recibir un tratamiento adaptado a su enfermedad y necesidades individuales. Asimismo, necesitará de un seguimiento médico regular que controle sus niveles de hierro, niveles de hemoglobina, hematocrito, etc., en definitiva, todos los parámetros afectados por la talasemia.
DIME DE DÓNDE VIENES Y TE DIRÉ QUE TALASEMIA TIENES
La distribución geográfica de las talasemias no es uniforme. Así, por ejemplo, la incidencia de estas anemias es mayor en zonas de África en las que la malaria es endémica. Esto es debido a que la hemoglobina defectuosa provoca un efecto protector frente al parásito responsable de dicha infección.
Por su parte, en la región mediterránea predominan las beta-talasemias, mientras que en Oriente Medio y Asia son más frecuentes, predominando las alfa-talasemias.
Por tanto, pertenecer a una determinada región o tener ascendencia de una zona en la que es mayor la incidencia para las talasemias hace más probable padecerlas o ser portadores de las mutaciones asociadas.
Asimismo, también conlleva una mayor gravedad, para el mismo tipo de talasemia, si el paciente es una mujer a causa de la pérdida de sangre durante la menstruación, que agravaría el estado anémico.
Ángela M. Martín Sevilla, Licenciada en Biología.
REFERENCIAS:
-Peña Olivar, I. (2016). Síndrome talasémico. Revisión Bibliográfica.