
EXOMA DIRIGIDO Y ESTUDIOS DE COSEGREGACIÓN, EL EQUIPO PERFECTO EN DIAGNÓSTICO GENÉTICO: CASO CLÍNICO
Introducción
En el mundo clínico es cada vez más común solicitar un exoma clínico dirigido a un fenotipo específico de un paciente, una herramienta que permite estudiar los exones, es decir, las secuencias codificantes, de un conjunto de genes que podrían guardar relación con la clínica de dicho paciente.
Una vez determinadas las diferentes variantes genéticas que presenta un paciente, y que podrían guardar relación con su clínica, entran en juego los estudios de cosegregación y segregación, que permiten, mediante el estudio de presencia o ausencia de esas variantes en progenitores u otros familiares del paciente, dilucidar si una determinada variante guarda o no, verdaderamente, relación con la clínica del paciente.
Caso clínico: motivo de consulta, antecedentes y exploración física

Para poder entender cómo estas dos herramientas de estudio genético forman una combinación perfecta en diagnóstico clínico, se presenta el caso de un paciente varón de 10 años con retraso psicomotor no filiado por microcefalia, retraso del crecimiento y del desarrollo, que presentaba además dilatación de las astas posteriores y un cuerpo calloso adelgazado, visto por resonancia magnética nuclear (RMN), según informe médico facilitado. El paciente no presentaba rasgos dismórficos característicos y respondía a las órdenes de manera adecuada, aunque sus capacidades de aprendizaje eran inferiores a la media. Junto con ello, se le había realizado previamente un array de hibridación genómica comparativa (CGH, por sus siglas en inglés) con resultado negativo, en el que no detectaron alteraciones de ganancia o pérdida de material genético en el genoma del paciente. En base al fenotipo del paciente, se solicitó un estudio de exoma clínico dirigido a retraso psicomotor y microcefalia.
Exoma clínico dirigido: secuenciación y resultados obtenidos
Tras analizar las variantes de significado clínico patogénico y probablemente patogénico en más de 23.000 genes, así como las variantes de significado clínico incierto en genes relacionados con el fenotipo del paciente, se seleccionaron ocho variantes puntuales en genes que podrían guardar relación con la clínica. De todas estas variantes, nos llamó la atención una descrita como patogénica en bases de datos genéticos como ClinVar y clasificada como tal según los criterios de la American College of Medical Genetics (ACMG), en el gen TUBG1. Se trataba de una variante puntual en la posición nucleotídica 1022 en la que se produce el cambio de una guanina por una adenina (c.1022G>A), dando como consecuencia el cambio aminoacídico de un residuo de arginina por uno de glutamina en la posición 341 de la proteína final (p.Arg341Gln).
Alteraciones en TUBG1 se relacionan con Displasia Cortical, Compleja, Con Otras Malformaciones Cerebrales 4 (OMIM: 615412), un trastorno de migración neuronal aberrante con un patrón de herencia
autosómico dominante, es decir, en el que solo es necesaria la presencia de un alelo mutado (estado heterocigoto) para causar sintomatología. Entre las características clínicas de esta patología se encuentran microcefalia y cuerpo calloso adelgazado, dos rasgos fenotípicos característicos que presenta nuestro paciente, junto con convulsiones de inicio temprano y diversas malformaciones del desarrollo cortical.
Tras consultar bibliografía y bases de datos disponibles, como GeneReviews, en busca de información relevante sobre el gen, nos llamó la atención que el 95% de pacientes afectos en los que variantes patogénicas en TUBG1 estaban involucradas en la presencia de clínica, presentaban dichas variantes causales de novo, por lo que era raro que una persona afecta presentara un progenitor también afecto.
Al tratarse de una patología con herencia autosómica dominante, la variante debería haber sido, a excepción de variantes de novo, transmitida al afecto por alguno de los progenitores. En este punto entran en juego los estudios de segregación y cosegregación, ya que para este patrón de herencia el hecho de que un progenitor presente dicha variante y no presente clínica, es decir, sea asintomático, permite descartar automáticamente esa variante como causal del fenotipo del paciente.
Estudio de segregación/cosegregación: secuenciación de los progenitores y resultados obtenidos
Tras el exoma dirigido, el clínico solicitó un estudio de segregación familiar de las variantes patogénicas encontradas y de cosegregación de las variantes inciertas, en los progenitores del afecto, con el fin de descubrir la verdadera relevancia clínica de las variantes encontradas.
Al estudiar en ambos progenitores, sin antecedentes personales de interés, las variantes seleccionadas en el exoma clínico dirigido, se obtuvo como resultado que todas las variantes, excepto la encontrada en el gen TUBG1, habían sido transmitidas al afecto por alguno de sus progenitores, por lo que serían descartadas aquellas ligadas a patologías de herencia autosómica dominante y no sería descartado el estado de portador en patologías de herencia autosómica recesiva, donde es necesaria la presencia de dos alelos mutados para desarrollar sintomatología.
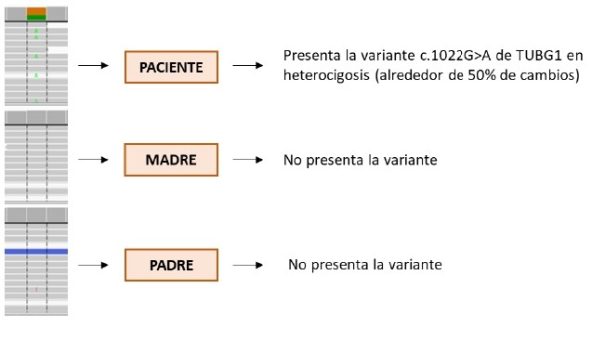
Al no presentar ninguno de los progenitores la variante mencionada anteriormente en el gen TUBG1, pudimos concluir que había aparecido de novo en el paciente, lo cual concordaba con la información revisada en las bases de datos. Al ser una variante patogénica de novo, podría causar clínica y explicar parte del fenotipo del paciente, así como por qué sus progenitores son asintomáticos, es decir, no existen antecedentes familiares. Al tratarse de una variante de novo en la línea germinal de alguno de los progenitores, es decir en la formación de los gametos, o después de la fecundación en el cigoto, el paciente presenta dicha variante en cada célula de su cuerpo y podrá transmitirla a su descendencia.
Conclusión
Por tanto, pudimos concluir que los resultados combinados de los estudios de exoma clínico y segregación/cosegregación permitieron encontrar una variante de significado clínico patogénico, según criterios de la ACMG, que había aparecido de novo en el paciente y que podría explicar parte de su clínica, especialmente los rasgos de microcefalia y cuerpo calloso adelgazado.
Autor: Gonzalo Valentín Lancha, graduado en Bioquímica. Analista Genético en Lorgen GP.